Дизметаболические нефропатииСтраница 3
Нередко у детей встречается УРАТНАЯ НЕФРОПАТИЯ, которая характеризуется интерстициальным нефритом и развитием нефролииаза, возникающим при нарушении пуринового обмена с накоплением и избыточным выведением через почки мочевой кислоты и ее солей (ура-тов). По сводным данным, уратные нефропатии являются причиной формирования нефро- и уролитиаза у 1,3—7,6% детей с мочекаменной болезнью.
В зависимости от механизмов происхождения различают первичные и вторичные уратные нефропатии. Первичные нефропатии возникают вследствие наследственно обусловленного дефекта метаболизма мочевой кислоты, а вторичные являются осложнением эритремии, миелом-ной болезни, хронической гемолитической анемии, лекарственной терапии тиазидовыми диуретиками, цитостатиками, салицилатами и др.
Нарушения пуринового обмена, приводящие к уратным нефропати-ям, довольно сложны, и выявляется несколько механизмов, которые лежат в их основе. В частности, уратные нефропатии возникают в результате (рис. 85):
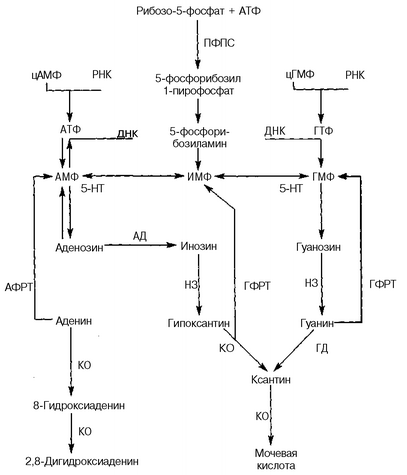
Рис. 85. Схема метаболизма пуринов в норме и патологии: КО — ксантиноксидаза, ГФРТ — гипоксантин-гуанин-фосфорибозил трансфераза, АФРТ— аденин-фосфорибозил трансфераза, ПФПС — пурин-фосфорибозил-пирофосфат синтетаза, ГД — гуанин-дезаминаза, НЗ — нуклеозидаза, 5-ПТ — 5-нуклеотидаза, АД — аденозин-дезаминаза
а) высокой активности пурин-фосфорибозил-пирофосфат синтетазы (ПФПС), обеспечивающей перенос пирофосфатной группы с АТФ на рибозо-5-фосфат с образованием 5-фосфорибозил-I-пирофосфата, который служит основой образования инозинмонофосфата и в последующем — мочевой кислоты. Этот механизм является наследственно обусловленным (описано более 20 семей) и связан с дефектом генов, локализованных в Х-хро-мосоме;
б) низкой активности фермента аденин-фосфорибозил трансферазы (АФРТ), который обеспечивает превращение аденина в аденозинмоно-фосфат, что способствует накоплению 2,8-дигидроксиаденина, обладающего нефротоксичностью и формирующего мочевые камни. Указанный дефект наследуется по аутосомно-рецессивному типу и обусловлен дефектными генами, которые находятся в 16-й хромосоме;
в) отсутствия фермента гипоксантин-гуанин-фосфорибозил трансфе-разы (ГФРТ), который катализирует превращение гипоксантина в ино-зинмонофосфат и гуанина — в гуанозинмонофосфат. Это ведет к гиперпродукции ксантина (из гипоксантина и гуанина) и мочевой кислоты (синдром Леша—Нихана, по имени авторов, описавших его в 1964 году). Указанный генетический дефект сцеплен с полом (болеют только мальчики), поскольку дефектные гены локализуются в Х-хромосоме.
Одним из проявлений нарушения пуринового обмена является ксан-тинурия, которая также часто ведет к нефро- и уролитиазу. Это патологическое состояние является одним из патогенетических вариантов уратных нефропатий. Ксантинурия может иметь наследственное и приобретенное происхождение. Наследственная ксантинурия обусловлена врожденным дефектом активности фермента ксантиноксидазы, который катализирует превращение гипоксантина в ксантин и далее — в мочевую кислоту, а также аденина — в 8-гидроксиаденин и 2,8-дигидрокси-аденин. Дефектные гены, контролирующие активность ксантиноксида-зы, локализуются во 2-й хромосоме. Приобретенная низкая активность ксантиноксидазы может быть обусловлена длительным приемом алло-пуринола, например у больным синдромом Леша—Нихана.
Выраженный и стойкий дефицит указанных выше энзимов приводит к повышенной продукции пуриновых оснований и гиперурикемии. Повышению биосинтеза пуриновых оснований способствует также избыточное питание. Накопление кристаллов мочевой кислоты в организме ведет к их отложению в интерстициальной ткани мозгового слоя, каналь-цевой системе, особенно в области петель нефрона, чашечно-лоханочном аппарате. При этом стенка канальцев повреждается, и выпавшие кристаллы проникают в интерстициальную ткань, накапливаются в ней и вызывают развитие интерстициального нефрита. При кислой реакции мочи могут формироваться уратные камни. Следствием высокой степени насыщения пуринами является уменьшение почек в размерах.