Дизметаболические нефропатииСтраница 1
Дизметаболические нефропатий — это группа заболеваний детского организма, которая характеризуется интерстициальным процессом с поражением канальцев почек в результате нарушения обмена веществ.
Эта группа заболеваний встречается очень часто в детском возрасте. По имеющимся данным, у 31,4% детей дошкольного возраста наблюдается оксалатно-фосфатно-кальциевая кристаллурия.
К дизметаболическим нефропатиям относят заболевания, связанные с нарушениями метаболизма щавелевой кислоты и кальция, пуринового обмена, метаболизма аминокислот и др.
ОКСАЛАТНАЯ НЕФРОПАТИЯ — патологическое состояние организма, возникающее при нарушении обмена щавелевой кислоты и характеризующееся поражением почек в результате отложения оксалата кальция в канальцах и интерстиции.
В зависимости от механизмов происхождения оксалатной нефропа-тий различают: первичные (наследственные) и вторичные гипероксалу-рии.
Первичная гипероксалурия является наследственно обусловленным заболеванием, связанным с отсутствием ферментов обмена глиоксило-вой кислоты. Заболевание наследуется по аутосомно-рецессивному типу, однако описаны случаи доминантного типа наследования.
В зависимости от патогенеза первичной гипероксалурии различают два типы заболевания:
I тип — длительное время считалось, что этот тип первичной гипер-оксалурии обусловлен недостаточностью активности фермента альфа-ке-тоглютаратглиоксилаткарболигазы, кофактором которого является фос-форилированная форма витамина В,. В связи с этим нарушается процесс метаболизации глиоксилата в кетоадипиновую кислоту. Однако проведенные в последние годы исследования убедительно показали, что I тип первичной гипероксалурии обусловлен низкой активностью фермента ала-нин-глиоксилатаминотрансферазы в клетках печени, который обеспечивает процессы трансаминирования глиоксилата. Указанный дефицит фермента ведет к накоплению и повышенному выделению с мочой гликолата, глиоксилата и солей щавелевой кислоты (оксалата кальция) (рис. 84).
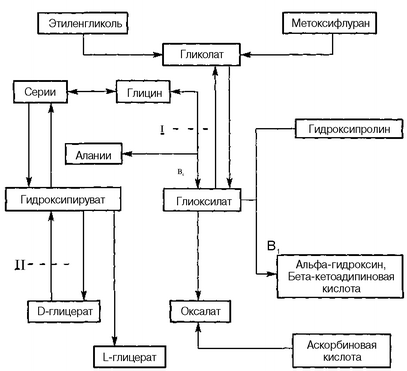
Рис. 84. Схема метаболизма оксалатов в норме и при оксалатной нефропатии
II тип связан с отсутствием или недостаточностью фермента В-гли-цератдегидрогеназы, который обеспечивает превращение гидроксипиру-вата в В-глицерат (рис. 84). В результате наблюдается избыточное образование гидроксипирувата, глиоксилата, оксалата кальция и Ь-глицерата и повышенная экскреция указанных веществ с мочой. При этом типе первичной гипероксалурии резко снижается или полностью прекращается экскреция с мочой гликолата.
В результате указанных дефектов наблюдается отложение кристаллов оксалата кальция, прежде всего — в проксимальных извитых канальцах нефронов и интерстиции почек, что сопровождается дегенерацией эпителия канальцев, утолщением интимы почечных артериол и сужением их просвета. По мере прогрессирования заболевания отложение оксалата происходит в головном мозге, в костях и хрящах, стенках сосудов, лимфатических узлах, ряде эндокринных желез, селезенке и других органах. Процессы кальцификации приобретают генерализованный характер, вследствие чего возникают нарушения функций органов и систем, нефролитиаз, гидронефроз, интерстициальный нефрит, ведущие к развитию хронической почечной недостаточности.
Вторичная гипероксалурия — патология обмена, обусловленная гаст-роинтестинальной гиперабсорбцией щавелевой кислоты из богатых ею пищевых продуктов, а также экзо- или эндогенным дефицитом витаминов В, или В6, являющихся кофакторами ферментов, осуществляющих метаболизм глиоксилата. Локальное образование оксалатов в почках возможно в связи с разрушением мембранных фосфолипидов на фоне ишемии почек. Морфологически выявляют деструкцию щеточных каемок в канальцах нефрона, лимфогистиоцитарную инфильтрацию в ин-терстиции почек.
Основные клинические симптомы первичной гипероксалурии обнаруживаются в раннем возрасте. Заболевание обычно проявляется у двух третей детей в возрасте 3—5 лет. Первичная гипероксалурия составляет до 2% всех случаев уролитиаза у детей. Мальчики болеют несколько чаще девочек.