Наследственный нефритСтраница 2
При аутосомно-рецессивном варианте синдрома Олпорта обнаружены мутации в локусе гена COL4A3 и COL4A4 на 2-й хромосоме. В настоящее время известно около 10 вариантов мутаций указанных генов.
Одновременная мутация, обусловленная делецией, в генах CoL4A5 и CoL4A6, кодирующих синтез альфа-5 и альфа-6 цепей коллагена IV типа, ответственна за возникновение сцепленного с Х-хромосомой синдрома Олпорта, течение которого сочетается с лейомиоматозом пищевода и гениталий.
Недавно описан новый вариант синдрома Олпорта у 4 членов одной семьи (мать, два сына и дочь), связанный с делецией в области Хд22.3-д23 хромосомы рецессивных генов, окружающих ген COL4A5. Особенностью клинических проявлений этого варианта является то, что помимо нефрита выявляется задержка психического развития, дисморфное лицо с гипоплазией средней части и элиптоцитозом. При этом элиптоцитоз не связан с нарушением мембранных протеинов эритроцитов. Задержка психического развития у больных с данным вариантом синдрома Олпорта обусловлена, по-видимому, отсутствием энзима ацил-Со-синтетазы ACS4 и связанного с ним специфического мозгового ACS3 протеина.
Таким образом, в результате мутаций нарушается процесс сборки молекулы коллагена IV типа, происходит изменение трехспиральной структуры коллагена, который состоит из двух альфа-1 цепей (IV) и одной альфа-2 цепи (IV), а также содержит незначительное количество альфа-3 (IV), альфа-4 (IV) и альфа-5 (IV) цепи. Сеть из молекул коллагена IV типа в соединении с другими белками экстраклеточного матрик-са (ламинин, протеогликаны, энтактин) является структурным каркасом гломерулярного фильтрующего барьера, который состоит из клеток эпителия, соединительной ткани и собственно базальных мембран. На ранних стадиях синдрома Олпорта (рис. 77) дефект этой сети определяет истончение и ломкость гломерулярных базальных мембран (ГБМ), что клинически проявляется гематурией и/или лротеинурией, а на поздних стадиях — способствует нарушению проницаемости и утолщению гло-мерулярных базальных мембран, проявляясь нарастанием протеинурии и снижением функции почек.
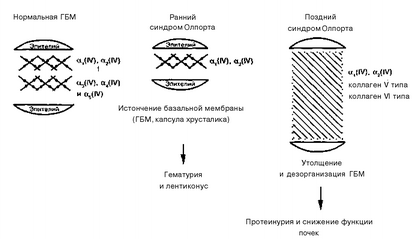
Рис. 77. Основные этапы изменения базальных мембран гломерул при наследственном нефрите (Ф.Д. Цаликова, 1998)
Следует отметить, что мутации гена CoL4A5 при Х-сцепленном синдроме Олпорта очень часто ассоциируются с отсутствием альфа-3, альфа-4, алъфа-5 и альфа-6 цепей IV типа коллагена в базальных мембранах, тогда как возрастает количество альфа-1 и альфа-2 цепей в ГБМ. Однако механизмы этого явления неизвестны. В культуре фибробластов кожи больных Х-сцепленным синдромом Олпорта показано, что это происходит в результате только посттранскрипционных событий, поскольку уровень мРНК для альфа-1 и альфа-6 цепей IV типа коллагена не отличается от здоровых людей.
МОРФОЛОГИЯ. Указанные процессы приводят к изменению морфологии гломерул. В качестве основных светооптических признаков синдрома Олпорта признаны меньшие, чем в норме, капиллярные петли и гломерулы, похожие на «фетальные». Диффузная мезангиальная пролиферация, сегментарный или глобальный гломерулосклероз, перигломе-рулярный фиброз, атрофия канальцев, интерстициальный фиброз, часто в интерстиции выявляются «вспененные» клетки, но они неспецифичны. При электронной микроскопии выявляются ультраструктурные изменения гломерулярных базальных мембран, которые характеризуются истончением (особенно lamina densa), расслоением на 2 слоя или более и наличием электронно-плотных гранул диаметром около 40 нм. Дефекты в гломерулярных базальных мембранах могут иметь форму так называемых «баскетбольных корзин», изменение их толщины характерно, но не пато-гномонично для синдрома Олпорта. С помощью иммунофлюоресценции каких-либо изменений выявить не удается.
КЛАССИФИКАЦИЯ. В настоящее время используется классификация наследственного нефрита, предложенная М. Gubber и R. Habib (1988), согласно которой выделяют 3 варианта наследственного нефрита.