КардиомиопатииСтраница 1
Термин «кардиомиопатия» впервые был использован в 1957 году для обозначения группы заболеваний миокарда неизвестной этиологии. В 1972 году было дано следующее определение кардиомиопатий: «Кардиомиопатия — это острое, подострое или хроническое поражение сердечной мышцы неизвестной или неясной этиологии, часто сочетающееся с поражением эндокарда, иногда и перикарда». Это определение принято экспертами ВОЗ как основополагающее. Эта патология не вызвана ревматизмом, кардитом, диффузными заболеваниями соединительной ткани, хотя в последние десятилетия ряд авторов выделяют первичные и вторичные (или симптоматические) кардиомиопатий, которые являются атрибутом ряда заболеваний: при нарушении обмена веществ (гликогеноз, гемохроматоз, парфериновая болезнь), эндокринопатиях (микседема), при нейромышечных дистрофиях, токсических состояниях (наркомания и др.).
Согласно классификации ВОЗ, кардиомиопатий классифицируют по ведущим гемодинамическим или анатомическим признакам. В соответствии с этим различают три формы кардиомиопатий: 1) дилатационную (или застойную); 2) гипертрофическую; 3) рестриктивную (рис. 28).
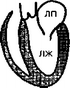
Норма
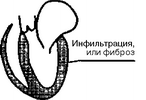
Рестрикгивная кардиомиопатия
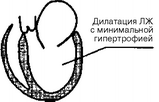
Дилатационная кардиомиопатия
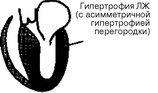
Гипертрофическая кардиомиопатия
Рис. 28. Анатомические изменения при кардиомиопатиях: ЛП — левое предсердие; ЛЖ — левый желудочек
ДИЛАТАЦИОННАЯ КАРДИОМИОПАТИЯ - это тяжелое заболевание миокарда, характеризующееся расширением полостей сердца, снижением его сократительной способности, развитием сердечной недостаточности, нарушением ритма сердца, тромбоэмболиями. Диагноз дилатационной кардиомиопатии устанавливают при наличии дилатации и систолической дисфункции левого желудочка и отсутствии врожденных пороков сердца, коронарной, клапанной или перикардиальной патологии, а также поражения сердца при гипертензии.
ЭПИДЕМИОЛОГИЯ. Действительная распространенность дилатационной кардиомиопатии не установлена в связи с недостаточной изученностью заболевания. Эпидемиологические данные о дилатаиионной кардиомиопатии свидетельствуют о том, что ее распространенность в разных странах мира составляет от 1 до 10 случаев на 100 000 населения.
ЭТИОЛОГИЯ. Наиболее вероятным представляется наличие нескольких специфических причин данного состояния. Дилатационная кардиомиопатия может носить семейный или предположительно генетический характер.
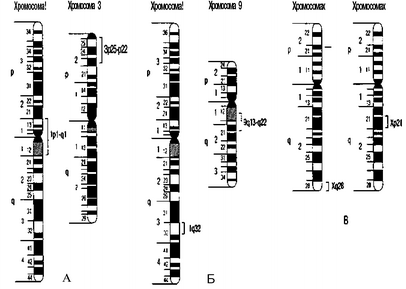
Рис. 29. Картирование генов у больных с дилатационной кардиомиопатией: А — при аутосомно-доминантном наследовании с сопутствующими дефектами; Б — при аутосомно-доминантном наследовании (без других дефектов); В — при Х-сцепленном наследовании
Семейная форма дилатационной кардиомиопатии встречается редко, составляя 6—9% всех случаев заболевания. Описаны случаи аутосомно-доминантного и рецессивного наследования, а также известна форма заболевания, сцепленная с Х-хромосомой. При картировании генов (рис. 29) у больных дилатационной кардиомиопатией с нарушениями функции проводимости дефектные гены выявлены в коротком плече хромосомы 3 (Зр25-р22) и в хромосоме 1 (1р1-д1), а при «чистой» форме заболевания — в хромосомах 1 (1д32) и 9 (9д13-д22). В случае Х-сцепленной формы (рис. 26) дилатационной кардиомиопатии мутантный ген локализуется в коротком плече Х-хромосомы (Хр21) или на длинном плече Х-хромосомы—Хд28 (при синдроме Барта).